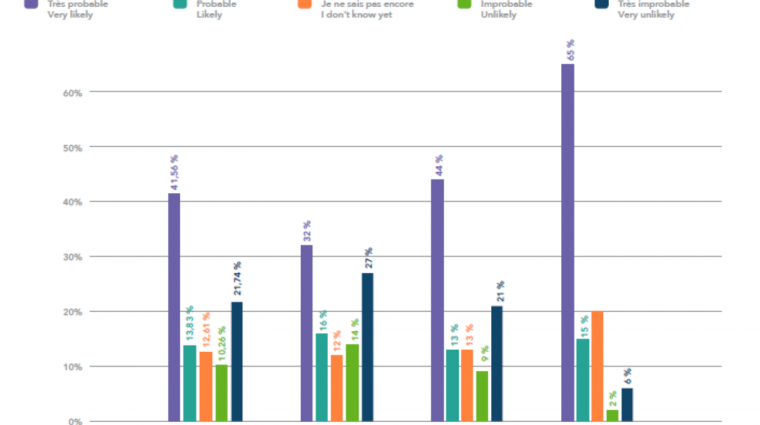
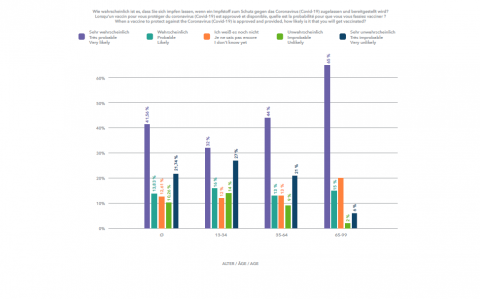
Am 10. Januar 2020 veröffentlichte ein Team chinesischer Wissenschaftler die Erbgutsequenz des neuen Coronavirus Sars-CoV-2 – einen Tag bevor China den ersten Corona-Todesfall meldete. Nicht mal ein Jahr später stehen mehrere hochwirksame Impfstoffe vor der Zulassung. Diese Geschwindigkeit schlägt alle Rekorde. Typischerweise nimmt die Entwicklung eines Impfstoffs etwa 10 bis 15 Jahre in Anspruch. Selbst in schnellen Fällen dauert es vier bis fünf Jahre. Ein Beispiel ist das Mumps-Vakzin, das in den 1960ern innerhalb von nur vier Jahren entwickelt worden ist. Und nach dem Ebola-Ausbruch in West Afrika im Jahr 2014 dauerte es fünf Jahre bis zur Zulassung eines Impfstoffs.
Weshalb dauert dies normalerweise so lange?
- Allein schon die Vorstudien, etwa zur Klärung der Frage, gegen welchen Teil des Virus eine Impfung Schutz aufbauen muss, können sonst Jahre in Anspruch nehmen.
- Präklinischen Studien, in denen möglicherweise wirksame Stoffe im Tierversuch getestet werden, und in denen Daten zur Toxikologie erhoben werden, dauern zwei bis vier Jahre.
- Danach folgen klinische Studien, die gestaffelt in die Phasen I, II und III, eine immer höhere Anzahl an Probanden umfassen, und die weitere fünf bis sieben Jahre andauern können.
- Erst am Ende der gesamten Studien erhalten die Behörden die Resultate zur Begutachtung – ein Prozess, der gut und gern noch einmal ein bis zwei Jahre in Anspruch nehmen kann.
Dass der Covid-19-Impfstoff nun derart rasant entsteht, weckt Befürchtungen: Wurde die Sicherheit des Impfstoffs der Geschwindigkeit geopfert? Hat man bei der Überprüfung der Risiken Abkürzungen genommen?
Für jede einzelne Phase der Impfstoffherstellung gibt es Gründe, weshalb diese schneller möglich war als sonst üblich. Wir gehen auf die einzelnen Punkte ein. Zu Anfang, für alle Eiligen, eine Kurzfassung der Hauptgründe. Danach gehen wir dann zu den einzelnen Punkten mehr in die Tiefe.
Kurzfassung: Hauptgründe, weshalb es bei der Entwicklung des Sars-CoV-2-Impfstoffs so schnell ging
Weshalb war die präklinische Phase kürzer?
- Beim Ausbruch von SARS (2002) und MERS (2012), beides Coronaviren, wurde bereits an Impfstoffkandidaten geforscht. Die Erkenntnisse von damals konnten erfolgreich auf Sars-CoV-2 (auch ein Coronavirus) angewendet werden. Die Forscher mussten nicht bei Null anfangen. Dies hat viel Zeit gespart.
- Ein bisschen Glück spielt auch eine Rolle: Gegen Coronaviren lassen sich relativ einfach Impfstoffe entwickeln. Das ist nicht bei jedem Virus so.
Weshalb waren die klinischen Phasen kürzer?
- Damit Phase 3 schnell vonstattengeht, müssen sich ausreichend Probanden mit der Krankheit infizieren, um Aussagen über die Wirksamkeit des Impfstoffs treffen zu können. Die vergleichsweise hohe Anzahl an Studienteilnehmern sowie die zurzeit hohe Prävalenz in der Bevölkerung bewirkte, dass sich schnell ausreichend Teilnehmer infizierten, und also vor allem diese Phase schneller als üblich voranging.
- Bei den Impfstoffkandidaten von Pfizer/Biontech und Moderna kam es während der klinischen Studien zu keinen Komplikationen, welche die Prozesse verlängert hätten. Dies war aber z.B. beim Impfstoff von Astra Zeneca/Oxford anders, weshalb dieser Impfstoff – obwohl er anfangs die Nase vorn hatte – nun nicht mehr in der Pole Position ist.
- Anstatt zuerst Phase 1, dann 2 und dann 3 separat hintereinander zu schalten wurden die einzelnen Phasen teilweise überlappt.
Statistik und Ressourcen: Viele Projekte gleichzeitig – viele Mittel die zur Verfügung gestellt werden
- Die finanziellen Mittel, die für die Impfstoffentwicklung gegen Covid-19 zur Verfügung gestellt wurden, sowie die Kooperation zwischen privaten und öffentlichen Partnern, ist einzigartig. Es wurde ausserdem zeitgleich an vielen verschiedenen Impfstoffkandidaten gearbeitet. Diese beiden Tatsachen haben einen Effekt auf die Qualität und die Quantität der Impfstoffkandidaten (aktuell über 200 Impfstoffkandidaten in klinischen Studien): Dadurch erhöht sich schlicht und ergreifend die Wahrscheinlichkeit, dass ein oder mehrere Impfstoffkandidaten gut und reibungslos funktionieren.
Entwicklungen in der Forschung
- Seit nunmehr über 25 Jahren wird an mRNA-Impfstoffen geforscht. Die Technologie war bereits weit fortgeschritten und ist nun einsatzbereit. Die Mittel, die in die Impfstoffentwicklung gesteckt wurden, haben höchstwahrscheinlich dazu beigetragen, dass es nun "auf die letzten Meter" nochmals schneller ging, aber es kommt in der Forschung immer wieder vor, dass Technologien ab einem gewissen Punkt einsatzbereit sind und sich ab dann durchsetzen. Die mRNA-Impfstoff-Technologie ist eine solche vielversprechende Technologie, die in Zukunft häufig zum Einsatz kommen könnte.
Weshalb gingen die Zulassungsprozeduren schneller?
- Behörden setzen viel Ressourcen frei, um zu kooperieren. Anstatt z.B. alle Studien zuerst abzuschliessen und dann erst die Genehmigungsprozedur zu starten, wird auf sogenannte Rolling Reviews zurückgegriffen, wo Behörden bereits Einsicht in die Daten erhalten, während die Studien noch laufen.
Weshalb konnte die Massen-Produktion so schnell anlaufen?
- Die Impfstoffhersteller haben bereits die Massenproduktion anlaufen lassen, bevor ihre Impfstoffkandidaten genehmigt wurden. Hiermit sind sie ein finanzielles Risiko eingegangen.
Wie man sieht: Dass die Impfstoffentwicklung viel schneller ging als üblich, hat durchaus erklärbare Gründe. Es wurden aber - anders als bei z.B. dem russischen Impfstoff - alle Phasen der Sicherheits- und Wirksamkeitsprüfung eingehalten.
Ging die schnelle Entwicklung also auf Kosten der Sicherheit?
(Die nun folgenden Passagen wurden am 22. Dezember upgedated):
Wenn es im Fall vom Impfstoff von Biontech/Pfizer nach der Evaluation der EMA (der Europäischen Arzneimittel Agentur) geht: Nein. Dieser Europäischen zuständigen Behörde wurden alle Daten der präklinischen und klinischen Phasen vorgelegt. Sie hat, nach Durchsicht aller Daten, den Impfstoff zur Verwendung empfohlen. Dies bedeutet, dass sie die Chancen des Impfstoffs hoch und die Risiken als niedrig bewerten - und dass die Prozeduren eingehalten wurden und den gängigen Sicherheitsstandars entsprechen.
Dies bedeutet nicht, dass es zu gar keinen Nebenwirkungen kam. Die Risiken derselben wurden bloss nicht als hoch eingestuft. Das bedeutet auch nicht, dass zu diesem Zeitpunkt bereits über Langzeiteffekte oder seltene Nebenwirkungen befunden werden kann. Dies ist zurzeit noch gar nicht möglich. Über Langzeiteffekte kann erst nach einer gewissen Zeit befunden werden. Und seltene Nebenwirkungen können erst ermittelt werden, wenn sich sehr viele Menschen haben impfen lassen. Dies wird in der sogenannten Phase 4, nach der ersten Marktzulassung, getan.
Während Monaten/Jahren werden die Impfstoffe nun weiterhin auf ihre Sicherheit und Wirksamkeit geprüft. Kommt es während dieser Zeit zu schwerwiegenden Komplikationen, kann ein Impfstoff wieder vom Markt gezogen werden. Dies ist ein übliches Prozedere, das bei jedem Medikament und bei jedem Impfstoff so angewendet wird. Es geht auch gar nicht anders: Seltene Nebenwirkungen und Langzeitfolgen können nur auf diese Weise ermittelt werden. Aufgabe der EMA war, sicherzustellen, dass in den präklinischen und klinischen Phasen 1-3 alles korrekt und nach den Sicherheitsstandars ablief. Was nun in den nächsten Wochen und Monaten folgt, muss aber weiterhin aufmerksam verfolgt werden.
All diese Ausführungen antworten vor allem auf die Frage, weshalb es so schnell gehen konnte. Sie antworten auch auf die Frage, ob die gängigen Sicherheitsstandards bisher eingehalten wurden (Ja, zumindest sieht die EMA dies so). Diese Ausführungen gehen aber nicht im Detail auf die bisher bekannten Risiken und Nebenwirkungen der Covid-19 Impfstoffe ein. Dies haben wir jedoch in folgendem Artikel getan:
Da die einzelnen Aspekte zu Schnelligkeit der Entwicklung bisher nur oberflächig behandelt wurden, hier dann die einzelnen Punkte mit mehr Hintergrundinformationen.
«Die Forschenden mussten nicht bei Null anfangen»
Ein erstes wichtiges Element, dass so schnell ein Impfstoff entwickelt werden konnte, ist die zügige Entschlüsselung des Virusgenoms. Die Tatsache, dass die chinesischen Forscher die Gensequenz offen ins Internet gestellt haben, machte es Wissenschaftlern aus aller Welt möglich, sofort nach jenem Abschnitt im Erbgut zu fahnden, der nach einer Impfung für die Bildung von Antikörpern wichtig ist. Dabei kam es gut zupass, dass die Forschenden schon eine ziemlich gute Idee hatten, wonach sie suchen mussten.
Bereits beim Ausbruch des ersten SARS-Virus 2002 bis 2004 hatten präklinische Studien mit möglichen Impfstoffkandidaten stattgefunden. Auch beim MERS-Virus wurde die Vakzin-Entwicklung aufgenommen. Zwar gibt es bisher keinen Impfstoff gegen diese beiden Viren, doch die damaligen Impfstoffstudien waren nicht umsonst. Weil sie mit Coronaviren durchgeführt wurden, die dem Sars-CoV-2 sehr ähnlich sind, helfen sie Forschenden aber nun beim Design von Impfstoffkandidaten. Etwa bei der Identifikation von wichtigen Antigenen. Antigene sind Bestandteile des Virus, gegen die unser Immunsystem eine Abwehrreaktion dirigieren kann, also gegen die es Antikörper bilden kann. Zum Beispiel das Stachelprotein in der Virushülle, also den «Zacken in der Krone» von SARS-CoV-2.
So kam es, dass der amerikanische Impfstoffhersteller Moderna keine zehn Wochen nach der Veröffentlichung des Virusgenoms einen Impfstoffkandidaten für Phase-I klinische Studien bereit hatte. «Die Forschenden mussten nicht bei null anfangen», sagt Carlos Guzmán, Leiter der Abteilung Vakzinologie am Helmholtz-Zentrum für Infektionsforschung in Braunschweig.
Die verschiedenen Phasen der klinischen Studien liefen parallel ab
Auf diesem Vorsprung ausgeruht haben sich die Impfstoffhersteller aber nicht. Sie konnten weitere Zeit gutmachen, indem sie die verschiedenen Phasen der klinischen Studien parallel laufen liessen.
Hier ein Überblick-Artikel, wie die einzelnen Phasen der Impfstoffherstellung ablaufen:

Normalerweise begutachten nicht nur die Wissenschaftler, sondern vor allem auch die Geldgeber die Ergebnisse einer jeden Phase peinlich genau, bevor sie mit der nächsten Stufe weiter machen. So beginnt normalerweise die grosse und kostspielige Phase III, bei der ein Impfstoffkandidat an zehntausenden Probanden untersucht wird erst dann, wenn die Daten aus den vorhergehenden Phasen I und II, mit einigen Dutzend beziehungsweise Hunderten Probanden, vielversprechend sind. Sofern das Risiko für ein Scheitern gering ist, und, wenn es einen Markt für den Impfstoffkandidaten gibt.
Im Falle der Covid-19-Impfung haben die Entwickler die Zwischenergebnisse aber nicht abgewartet, sondern trotz ungewissen Ausgangs nicht nur weitere Studienphasen aufgezogen, sondern bereits die Produktion der Vakzine aufgebaut. «Es ist die Bereitschaft, trotz finanziellem Risiko fortzufahren, die die beschleunigte Entwicklung von Sars-CoV-2-Impfstoffkandidaten ermöglicht hat», schreibt Florian Kramer, Professor für Vakzinologie an der Icahn School of Medicine at Mount Sinai in einem kürzlich erschienenen Übersichtsartikel in der Fachzeitschrift Nature.
Das finanzielle Risiko trugen in diesem Fall aber nicht nur die Firmen, sondern auch private Investoren sowie öffentliche Geldgeber. Staaten unterstützen die verschiedenen Projekte mit Milliardenbeträgen. So erhielt Moderna fast 1 Milliarde Dollar an öffentlichen Geldern. Und die Europäische Kommission unterstütze das Projekt von Biontech/Pfizer mit 100 Millionen Euro.
Zusammenarbeit zwischen Firmen, öffentlichen Institution und freiwilligen Studienteilnehmern
Diese Zusammenarbeit zwischen öffentlichen Institutionen und grossen Pharmafirmen, die «public-private partnership», ist denn auch ein besonderes Kennzeichen der Coronapandemie. So arbeitet Moderna etwa mit dem US National Institute of Allergy and Infectious Disease zusammen und die Firma AstraZeneca mit der Universität Oxford. «Der Grad der Zusammenarbeit ist ohne Beispiel», sagt Impfexperte Guzmán. «Bei keinem anderen Impfstoff waren die Menschen so stark über jeden Schritt der Entwicklung informiert.» So hätten die verschiedenen Projekte voneinander lernen können, was die Entwicklungsprozesse noch weiter beschleunigt habe.
Hilfreich für die Schnelligkeit der Entwicklung war aber auch, dass sich durch das hohe öffentliche Interesse viele Freiwillige für Studien finden liessen, und dass die Covid-19-Krankheit während der Phase-III-Studien in der Bevölkerung relativ weit verbreitet war. Das heisst, dass ausreichend Menschen in der Kontrollgruppe der Phase-III-Studie erkrankten, wodurch Daten zur Wirksamkeit schneller vorlagen. Und: Man hat sich nicht nur auf einen Wirkstoff verlassen. Insgesamt sind 261 Kandidaten im Rennen – eine vergleichsweise hohe Zahl – was die Wahrscheinlichkeit auf einen schnellen Erfolg erhöht hat.
«Rolling review»: Begutachtung bereits während der laufenden Studie
Ein weiterer ausschlaggebender Faktor ist der Umgang der Behörden mit den Vakzinkandidaten. Normalerweise erhalten die Zulassungsbehörden, wie die Food and Drug Administration (FDA) in den USA oder die European Medicines Agency (EMA) in der EU ein Dossier zu Wirksamkeit und Sicherheit erst nach Beendigung aller Studien. Dies ist derzeit anders. Die Behörden haben Zugang zu den vorläufigen Daten aus den präklinischen und klinischen Studien und können diese parallel zu den noch laufenden Untersuchungen begutachten. Dieses Verfahren heisst «rolling review», also rollende Begutachtung. Der Prozess hatte sich bereits bei der Entwicklung des Ebola-Impfstoffs 2014 als tauglich erwiesen.
Die rollende Begutachtung bedeutet aber nicht, dass die Behörden weniger genau hinschauen. Vielmehr organisieren sie ihre Ressourcen und ihr Personal wegen der Dringlichkeit der Situation so, dass sie die Begutachtung in kürzerer Zeit schaffen. «So kann ein Impfstoff direkt nach der Beendigung der Studien zugelassen werden», sagt Vakzinologe Guzmán. Er wird dabei allerdings nur für Personengruppen erlaubt, an denen der Impfstoff getestet wurde. Beispielsweise wurden die jetzt entwickelten mRNA-Impfstoffe bisher nicht an Kindern untersucht, darum dürfen diese auch (noch) nicht geimpft werden.
Provisorische Zulassung
Der Prozess der Zulassung kann noch weiter verkürzt werden. «Basierend auf belastbaren vorläufigen Ergebnissen zur Sicherheit und Wirksamkeit kann eine provisorische Zulassung erfolgen», sagt Guzmán. Diese sogenannte «Conditional marketing authorisation», wie sie bei der zuständigen Behörde für Arzneimittelzulassung EMA in Europa heiβt, ist keine entgültige Marktzulassung, und sie wird nur unter folgenden Bedingungen erteilt:
- wenn durch die Impfung ungedeckte medizinische Bedürfnisse erfüllt werden
- wenn die Daten zeigen, dass die Vorteile des Impfstoffes die Risiken überwiegen
- es wahrscheinlich ist, dass der Antragsteller weitere, umfassendere Daten vorlegen kann, die bestätigen, dass der Nutzen weiterhin die Risiken überwiegt
- und der Nutzen einer sofortigen Verfügbarkeit der Impfung für die öffentliche Gesundheit gröβer ist als die Risiken, die aufgrund des Bedarfs an weiteren Daten enstehen würden.
Sowohl Biontech/Pfizer als auch Moderna haben in den vergangenen Tagen bei der EMA auf Basis ihrer Zwischenergebnisse aus Phase 3 einen Antrag für eine solche provisorische Marktzulassung beantragt. Sollten die Daten überzeugend genug sein, könnten die Gutachter der EMA innerhalb von Wochen eine provisorische Zulassung genehmigen.
Weitere Studien untersuchen Langzeiteffekte nach der Zulassung
Im Anschluss an die Zulassung (egal ob provisorisch oder definitiv) wird sowohl die Sicherheit als auch die Wirksamkeit von Impfstoffen in Phase-IV-Studien weiter über einen längeren Zeitraum überwacht. Hier können dann auch zusätzliche Alters- oder Bevölkerungsgruppen den Impfstoff verabreicht bekommen. Damit soll sichergestellt werden, dass auch sehr seltene oder geringfügige Nebenwirkungen oder eventuelle Langzeiteffekte aufgedeckt werden. Treten in dieser Phase signifikante Nebenwirkungen auf, kann eine Zulassung auch wieder zurückgezogen werden. So verzichtete z.B. 2001 das Pharmaunternehmen Baxter freiwillig auf die Zulassung eines Impfstoffs gegen durch Zeckenstiche verursachte Hirnhautentzündungen (FSME), nachdem mehrere Meldungen von sehr hohem Fieber gemeldet wurden. Der FSME-Impfstoff wurde danach angepasst und neu zugelassen. Das ist aber einer der sehr seltenen Fälle, in denen tatsächlich ein kausaler Zusammenhang zwischen Impfungen und Nebenwirkungen bewiesen werden konnte, die nach der Zulassung auftraten.
Der russische Impfstoff
Die Entwicklung des ersten Impfstoffs gegen SARS-CoV-2 in Russland verlief anders. Dort wurde ein Impfstoff («Sputnik V») zugelassen, ohne dass Daten aus Phase-III-Studien vorlagen. Er wurde nur in Studien der Phase I und II mit 76 Probanden getestet. Laut Hersteller deuten erste Zwischenergebnisse darauf hin, dass die Impfung in 91,4% der Fälle vor einer Infektion schützen soll (Ergebnisse von 28 Tagen nach der ersten Dosis, 7 Tage nach der zweiten).
Dieses Vorgehen der russischen Autoritäten ist so in Europa nicht möglich.
Werden die Sicherheitsstandards bei beschleunigter Impfstoffentwicklung beibehalten?
Bei der Zulassung in der EU aber erfüllen auch die schnell entwickelten Covid-19-Impfstoffe vergleichbare Sicherheitsstandards wie alle anderen Vakzine. Die Vorgaben, zum Beispiel was die Anzahl der nötigen Probanden angeht, waren dieselben, denen auch alle anderen Impfstoffe genügen müssen.
«Zum jetzigen Zeitpunkt lässt sich sagen, dass bisher keine schwerwiegenden Nebenwirkungen aufgetreten sind. Insofern scheinen die aktuellen Impfstoffe sicher zu sein», sagt Guzmán. Eine absolute Aussage lasse sich aber erst machen, wenn die Studien beendet seien und alle Informationen vorlägen. Dies beinhaltet auch die Ergebnisse von Phase IV Studien, bei der zusätzliche Alters- und Patientengruppen sowie sehr seltene Nebenwirkungen und mögliche Langzeiteffekte untersucht werden.
Was sind mögliche Nebenwirkungen der Covid-19 Impfstoffkandidaten?
Bei den Nebenwirkungen handelt es sich im Falle der am weitesten fortgeschrittenen mRNA-Impfstoffe von Moderna und Biontech/Pfizer um Müdigkeit, Kopfschmerzen, Muskelschmerzen, Schüttelfrost und vorrübergehende Schmerzen an der Injektionsstelle. «Das zeigt, dass diese Vakzine nicht so gut toleriert werden wie einige unsere herkömmlichen Impfungen etwa gegen Grippe oder Tetanus», so Guzmán. Aber trotzdem überwiege der Nutzen die Gefahren bei Covid-19 Risikogruppen.
Sehr seltene Nebenwirkungen, die etwa in einem von Zehntausend, einem von Hunderttausend oder in einem von einer Million Geimpften auftreten, wird man erst nach einer Impfstoffzulassung kennen. Dies gilt auch für alle anderen Vakzine. «Im Allgemeinen treten die meisten Nebenwirkungen innerhalb von zwei bis drei Monaten nach der letzten Impfdosis auf», sagt Guzmán.
Infobox
Bei dem Impfstoff Pandemrix von GlaxoSmithKline, der in der Schweinegrippepandemie von 2009 zugelassen wurde, kam es Monate nach der Zulassung und Impfung von Millionen Menschen weltweit in einigen Ländern gehäuft zu Berichten von Gesichtslähmungen bei Erwachsenen oder Narkolepsie bei Kindern. Für letztere lieferten zwei Forscherteams aus Finland Hinweise auf einen Zusammenhang mit der Pandemrix-Impfung. Eine definitive kausale Verbindung konnten sie allerdings nicht beweisen. Der Impfstoff wurde also weiter verabreicht. Nachdem diese Pandemie milder als erwartet verlief, wurde Pandemrix (und auch andere Impfstoffe gegen die Schweinegrippe ohne mögliche Nebenwirkungen) dann vom Markt gezogen. Der British Medical Journal deckte viel später auf, dass das Pharmaunternehmen GSK (Hersteller von Pandemrix) anscheinend schon früh Hinweise auf vermehrte Nebenwirkungen und sogar Todesfälle im Vergleich zu anderen Impfungen gegen die Schweinegrippe hatte, diese aber damals ignoriert habe. Diese Berichte kamen erst im Zuge von gerichtlichen Verfahren ans Licht.
In der aktuellen Coronavirus-Pandemie verläuft die Kommunikation von seiten der Pharmaindustrie bisher jedoch schneinbar transparenter ab – so wurden beispielsweise von den, durch mögliche schwere Nebenwirkungen bedingten, Unterbrechungen der Covid-19-Impfstoffstudie von AstraZeneca/Oxford University öffentlich berichtet. In beiden Fällen konnte hier jedoch kein Nachweis eines kausalen Zusammenhangs mit der Impfung bestätigt werden. Deshalb konnte die Studie dann jeweils wieder weiterlaufen.
Ein bisschen Glück und eine neue Impfstoff-Technologie
Dass es bei den Covid-19-Impfstoffen so schnell ging, hat aber auch etwas mit Glück zu tun. Denn nicht alle Viren lassen sich mit einer Impfung bekämpfen. Ein gutes – beziehungsweise schlechtes – Beispiel ist das HI-Virus, das Aids verursacht. Hier beissen sich Forscher seit Jahrzehnten die Zähne an der Entwicklung eines Impfstoffs aus. Zum einen, weil HIV schnell mutiert und zum anderen, weil die Forschenden das Immunsystem noch nicht dazu bringen konnten, neutralisierende Antikörper gegen das Virus zu bilden.
Kein Zufall aber ist es, dass mit den Covid-19-Impfstoffen auch eine neue Impfstoff-Technologie die Bühne betritt. Die mRNA-Technologie wird zwar schon lange erforscht, etwa als Impfung zur Bekämpfung von Krebserkrankungen. Doch bisher verliess man sich bei der Vakzin-Entwicklung lieber auf etablierte Technologien, etwa Impfung mit Eiweissen oder inaktivierten Erregern. Doch, während deren Herstellung sehr zeitaufwendig ist, lässt sich ein mRNA-Molekül innerhalb weniger Tage herstellen. Dieser Vorteil der Schnelligkeit überwog die Verlässlichkeit bewährter Technologien bei bisherigen Entwicklungen von Impfstoffkandidaten nicht und spielte daher eine geringere Rolle. Erst jetzt mit der weltweiten Pandemie mit vielen Opfern kommt der Vorteil zum Tragen – es muss alles schnell gehen.
Infobox
Ein mRNA-Impfstoff enthält Abschnitte der Boten-RN oder kurz mRNA des SARS-CoV-2, also dem Erbmaterial des neuen Coronavirus. Diese mRNA-Schnipsel werden in eine winzige Nanokapsel aus Lipiden gepackt, die dann als Impfung eingespritzt wird. Diese Lipidnanokapseln können dann von menschlichen Zellen aufgenommen werden, so dass die mRNA-Schnipsel von SARS-CoV-2 in die Zelle gelangen.
Menschliche Zellen (und Zellen anderer Lebewesen) enthalten auch mRNA. Sie sind «Übersetzungen» von Genen, also Teilen unserer DNA. Mithilfe dieser mRNA produzieren unsere Zelle bestimmte Proteine, die für die Funktion des Körpers gebraucht werden.
Nun sind unsere Zellen ziemlich «dumm»: sie können nicht zwischen eigener mRNA und fremder mRNA unterscheiden. Und produzieren deshalb mit Hilfe des Impfstoff-mRNA-Schnipsels ein Protein der Virusoberfläche.
Was passiert nun mit den SARS-CoV-2 Proteinen, die unsere Zellen hergestellt haben? Daraus entsteht kein ganzes Virus. Ein Geimpfter kann also von einer Impfung nicht an Covid-19 erkranken. Aber unser Immunsystem erkennt das Virusoberflächenprotein, und bildet eine Immunantwort dagegen. Das bedeutet: er produziert Antikörper und T-Zellen gegen SARS-CoV-2. Wenn wir dann später mit dem richtigen, vollständigen SARS-COV-2 in Kontakt kommen, erkennt unser Immunsystem das Virus direkt wieder und attackiert es, bevor es zu einem schweren Krankheitsverlauf kommen kann.
Die mRNA des Impfstoffs wird danach, genau wie auch unsere eigenen mRNAs, zerstört. mRNA ist also so etwas wie eine Snapchat-Nachricht. Sie ist nur vorrübergehend. Sie wird nicht in die menschliche DNA eingebaut und hat auch keinen Einfluss auf unser Erbmaterial, weder in Körperzellen noch in Zellen der Keimbahn (also Ei- oder Samenzellen).
Doch auch wenn die Entwicklung des Impfstoffs rasant verlief – eines wissen wir nicht: wie lange der Immunschutz anhält. Sicherlich einige Monate, soviel lässt sich aus den vorliegenden Daten ableiten. Und sicherlich schützen die am Weitesten fortgeschrittenen Impfstoffe vor der Covid-19-Krankheit. Doch wie lange eine Impfung gegen Covid-19 hält und ob sie in regelmässigen Abständen aufgefrischt werden muss, das wird sich in den kommenden Monaten und Jahren zeigen. Die Beobachtung der Reaktion des menschlichen Immunsystem – das ist ein Prozess, für den es keine Abkürzung gibt.
Autoren: Cornelia Eisenach (Scitec-Media), Michèle Weber (FNR), Jean-Paul Bertemes (FNR)